MPS patients are at a high risk of cervical cord injury, which may result in paralysis and sudden, premature death1
They may experience atlantoaxial instability,2–5 commonly associated with cervical cord compression and myelopathy
Recommendations
Use manual in-line stabilisation to prevent cervical spine injury1
Limit the degree of flexion/extension because of the possible laxity of ligaments (with or without odontoid dysplasia) and cervical stenosis1
Intubation: maintain the patient in a neutral position during intubation since the “sniff” position may not be possible. Use fibre-optic intubation or video laryngoscopy1
Maintain the rest of the spinal column in the neutral position, as compression may occur in other regions1
Neurophysiological monitoring is recommended for all patients undergoing prolonged procedures and/or procedures involving a spine or manipulation of the head1
Step 2: High Risk Anaesthesia
(both intubation and extubation)
Ensure sufficient space in the nose and mouth for passage of air
Risks
Respiratory failure and airway–related emergencies are a common cause of morbidity in MPS patients,1 especially during surgical interventions.6 Critical decreases in oxygen saturation may occur suddenly1
In an airway anaesthesia emergency, there may be less than 3–5 minutes to perform an emergency tracheostomy before permanent brain damage occurs7
ANY sedative can cause respiratory complications, severe hypoxaemia, and, consequently, neurological impairment1
Airway obstruction:
MPS patients may have obstructive sleep apnoea (OSA), increasing the risk of airway emergencies and chronic hypoxaemia8
Airway obstructions may cause difficulties with mask ventilation and intubation1
Temporomandibular joint contracture with difficulty opening the mouth, and accumulation of glycosaminoglycans (GAGs) in the tongue, oral pharynx, and larynx can impede access to the upper airway and identification of the glottis1. This may result in negative pressure pulmonary oedema, or an inability to ventilate/intubate1 or visualise the airway9
Serious complications may occur during extubation, including pulmonary oedema, and the need for re–intubation or emergency tracheostomy1
Recommendations
Have an otolaryngologist (ENT), preferably with MPS experience, readily available during any surgical procedure on MPS patients due to the high potential for an emergency tracheostomy1
Ensure the ENT is aware that performing an emergency tracheostomy is more difficult, has a higher risk and will take longer for a patient with MPS because of their shorter neck, thickened soft tissue, and the depth of their trachea1
Be prepared for alternative methods of intubation (e.g. fibre–optic intubation) if mask induction followed by oral tracheal intubation is unsuccessful1
An oral anxiolytic may reduce anxiety and increase the potential for successful fibre–optic intubation – but, if the patient falls asleep, he or she may desaturate to dangerous levels due to upper airway obstruction1
Have the pre–op nurse closely monitor oxygen saturation and call the anaesthesia team immediately if changes in oxygen saturation occur1
Provide supplemental O2 during intubation due to the potential for difficulty in ventilation and oxygenation1
Consider use of nitrous oxide to assist in placement of an intravenous catheter, followed by induction with midazolam or fentanyl (reversed by flumazenil and naloxone, if required)1
Consider placing the patient in the lateral position during induction phase if this improves their airway1
Use fibre–optic bronchoscopy for tracheal induction if patient has a diffcult airway5
Use of a laryngeal mask airway (LMA) or nasal airway has been found to improve ventilation during bronchoscopy1
Consider inserting a J–tipped guide–wire through the suction channel of the bronchoscope into the trachea, remove the bronchoscope and insert a ureteral dilator or airway exchange catheter over the wire, then advancing the endotracheal tube (ETT) over this to help guide it into the trachea5
Avoid use of muscle relaxants until endotracheal intubation is achieved1
Use an ETT that is 2–3 times smaller than expected based on age1
In order to increase oxygen delivery to the patient during fibre–optic bronchoscopy, consider advancing a short ETT into the contralateral nares to provide continuous O2 into hypopharynx. Also, attach O2 to the suction port of the bronchoscope and intermittently inject O2 from tip of fibre10
Ensure full reversal of the muscle relaxant and place a nasopharyngeal airway prior to extubation1
Perform extubation in an area with access to the full medical personnel required should the patient need immediate re–intubation or an emergency tracheostomy1
Step 3: Contact MPS Specialist
Emergency Tracheostomy may be necessary
Risks
Difficult intubations may result in injury to the glottis, stridor, infection or airway collapse1
Potential for chronic hypoxaemia due to obstructive sleep apnoea (OSA)1
Once MPS patients are extubated, re–intubation may not be possible, creating a potential emergency1
Recommendations
Extubation should not be performed until the patient is fully awake, a leak test has been carried out and there is adequate respiratory effort1
Always have an experienced otolaryngologist (ENT) or paediatric surgeon in the room during all surgical procedures on MPS patients due to the high potential for an emergency tracheostomy1
Step 4: Maintain Cardiac Monitoring
Risks
Significant cardiac manifestations are reported for MPS patients11,12
Cardiac valve disease is the most commonly reported cardiac manifestation in MPS patients,1,11,12 increasing the risk of mortality during surgical procedures1
Ischaemia and cardiac arrest due to hypotension may occur13
Recommendations
Perform an ECG to identify conduction abnormalities and signs of myocardial ischemia11
Perform an echocardiogram to identify cardiac valve regurgitation or stenosis as well as decreased function1
Monitor blood pressure using intra–arterial cannulae if surgery is lengthy or high risk1
Anaesthesia and MPS
Patients with mucopolysaccharidosis (MPS) who require surgery present a significant challenge to anaesthesiologists. The high prevalence of airway and respiratory system involvement combined with cardiovascular alterations, results in high anaesthetic risk for these patients.
MPS is a group of rare diseases, but because they are progressive, those with the condition may require numerous surgical interventions throughout their lives. For each of these interventions, a comprehensive pre-anaesthetic assessment should be carried out to thoroughly examine the patient’s current condition. Paying particular attention to how MPS has affected their airway and cardiorespiratory system will help to assess risks prior to surgery and design the best anaesthetic plan.
The most important aspects affecting both the pre-anaesthesia visit and intraoperative management of MPS patients are:
The airway management of MPS patients is perhaps the most complicated in paediatric anaesthesiology due to the accumulation of glycosaminoglycans (GAG) in the tissues. This can lead to anatomical alterations and obstructions at different levels of the airway, creating difficulties not only for orotracheal intubation, but also for face mask ventilation.
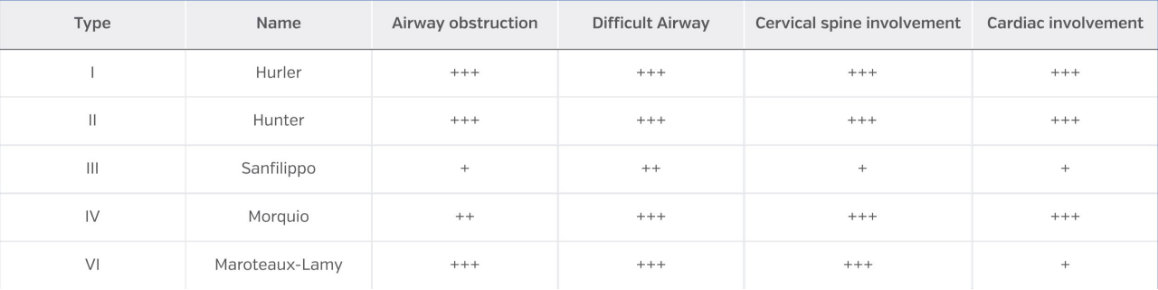
Airway involvement is greater in MPS types I, II, IV and VI, but less frequent in MPS III. With the implementation of enzyme replacement therapy, a later onset of these alterations has been observed which improves airway management in these patients. Even so, all patients with MPS should be considered as having a difficult airway until proven otherwise.
Bear in mind that airway involvement progresses throughout the patient’s life; therefore, the older the patient, the greater the likelihood of airway management difficulty. In addition, patients without intubation difficulty may develop the complication over time.
Airway disorders in MPS, by type
In MPS, airway disturbances can occur at any level:
Mouth
At the oral level, soft tissues may be affected. The presence of macroglossia is very common, with a very significant occupation of the oral cavity. This, added to hyperplasia of the palate and dental malposition, means that air permeability in this area can be compromised. Bone structures may also be affected, such as the temporomandibular joint (TMJ) and a hypoplastic mandibular ramus, resulting in limited mouth opening.
Pharynx
MPS patients usually have a flattened nasal bridge and adenoid hypertrophy, making them more susceptible to middle ear infections and chronic rhinorrhoea. They often have tonsillar hypertrophy and lymphoid tissue hypertrophy throughout the pharynx, making the mucosa thicker and more friable.
Larynx
The infiltration of GAGs at the laryngeal level alters the usual anatomy of the glottis. The epiglottis is usually large, and its anatomy is altered. The position of the glottis is more anterior and passage through the vocal cords is narrower, secondary to the infiltration of the arytenoids and other periglottic structures.
Trachea
Alterations at the tracheal level are also described. The most evident are subglottic tracheal strictures (although they can appear at any level of the trachea and bronchi, especially in more advanced stages of MPS) and tracheomalacia.
Skeletal
Several skeletal alterations are present at cranial and cervical spine level (flattened nasal bridge, mandibular and TMJ anomalies, hypoplasia or subluxation of C1-C2). Combined with the short neck characteristic of these patients and the possible compression of the upper cervical cord, these alterations can result in a limited mouth opening and impede the cervical extension manoeuvre used to facilitate airway management.
The involvement of the airways, lung parenchyma and chest wall in MPS patients creates a low compliance system, resulting in a restrictive pulmonary pattern.
At the pulmonary level, patients with MPS have a higher presence of mucus in the airway and are prone to recurrent airway infections. Bronchiectasis is also a frequent finding at the pulmonary level.
A restrictive pulmonary pattern can be created by extrapulmonary factors, including alterations in the rib cage (horizontalisation of the ribs, pectus carinatum) and at the level of the dorsal spine (scoliosis, kyphosis). Together with the pressure exerted on the diaphragm by the increase in size of the abdominal organs (hepatomegaly and splenomegaly), these alterations help to create a restrictive pattern at the pulmonary level.
In advanced stages of MPS, there is also an obstructive component due to the accumulation of deposits on the tracheal and bronchial walls.
In very advanced stages, when obstructive and restrictive patterns merge, and especially in patients with obstructive sleep apnoea-hypopnoea syndrome (OSAHS), pulmonary hypertension may develop due to hypoxaemia and chronic hypoventilation.
Obstructive sleep apnoea syndrome (OSAS) is common in patients with MPS and is a risk factor for perioperative respiratory complications. It is common for patients with OSAS to require non-invasive ventilation at home. If so, the use of their own non-invasive ventilation (NIV) ventilator during the perioperative period (especially in the immediate postoperative period) will be of great benefit.
Thus, in patients with MPS, a respiratory assessment prior to surgery is essential. This should include history and treatment of respiratory infections, severity of OSA, NIV ventilator use and, when necessary, an updated assessment by their pulmonologist for preoperative optimisation.
Cardiac involvement in patients with MPS are well described and defined, being more frequent in types I, II and VI than in types III and IV.
The most frequent diseases are: valvulopathies, cardiac dysfunction due to hypertrophic cardiomyopathy, electrical conduction disturbances and involvement of the coronary arteries.
The most frequent causes of death in MPS due to cardiac involvement are heart failure, sudden death due to arrhythmias and coronary occlusion.
Both the incidence and severity of cardiac problems increase over time due to the progressive nature of MPS. Despite this, the absence of cardiovascular signs or symptoms mean that the prevalence of heart disease is underestimated, even when it is present. This can lead to a complete cardiac evaluation being overlooked at certain times.
Valve involvement
Valve involvement is the most common manifestation of cardiac involvement in patients with MPS.
The most prevalent disease is valve insufficiency (greater than stenosis), and the most affected valve is the mitral valve, followed by the aortic valve. Therefore, left heart valves are more affected than right heart valves.
This valve dysfunction is a consequence of thickened valve tissue and a shortening of the (also thickened) papillary muscles, contraction of which is facilitated by the chordae tendineae. This results in limited valve motion and dysplastic valve structures.
Valvular regurgitation or stenosis can lead to left ventricular overload, ventricular dilatation, hypertrophy, and systolic and diastolic dysfunction.
In severe cases, valve replacement with mechanical or biological prosthesis can be performed.
Coronary artery involvement
Coronary artery occlusions are described in all types of MPS but are more frequent in types I and II.
The first involvement is a diffuse proliferation of the intima due to GAG deposits in the walls of the coronary arteries. Coronary arteriolar involvement and left ventricular apical aneurysms have also been reported.
Conduction alterations
The most frequently diagnosed arrhythmia in MPS is atrioventricular block in all its degrees, with complete atrioventricular block (in types II, III and VI) being common. This often requires pacemaker placement and is the main cause of sudden death.
Other vascular alterations
Thickening has also been reported in the abdominal and thoracic aortic artery walls as well as in renal arteries, leading to varying degrees of occlusion. In some patients, these deposits in the arterial walls may be the cause of systemic hypertension.
These alterations may lead to cardiac dysfunction requiring treatment with diuretics or vasodilators.
In addition to the airway, respiratory and cardiovascular system involvement that can affect MPS patients during the perioperative period, consideration should also be given to the following aspects to avoid complications.
Ophthalmological
The presence of exophthalmos is typical in MPS, as well as corneal lesions that may even require corneal transplantation. Retinal deposits and increased intraocular pressure may also be present.
To avoid eye injuries, special care must be taken both in intraoperative eye protection and face masks, and postoperative oxygen therapy masks.
Skeletal
Osteoarticular involvement can occur at every bone level. Avoiding positions during surgery that aggravate pain or cause nerve compression injuries is essential.
Pre-anaesthetic assessment with imaging tests of the cervical spine is vitally important to detect cervical compression or instability which can lead to changes in the vocal cords (arytenoids) and the spinal column.
Neurological
Anatomical alterations and osteoarticular involvement can lead to distortion of the peripheral nerve pathway, complications due to peripheral nerve compression and clinical complications due to spinal cord compression.
At the CNS level, hydrocephalus (which may require a ventriculoperitoneal shunt), certain degrees of brain atrophy and areas of demyelination may be found.
In patients with MPS, the anaesthetic plan should be carefully designed in advance and individually adapted to each patient. As for any anaesthetic procedure (but even more so in patients with MPS, due to the potential for additional complications), the main plan and any alternatives must be clear in case any difficulties should arise during the procedure.
Provided that the patient’s age and condition allow it, it is obviously preferable to avoid general anaesthesia.
Premedication
MPS patients will have endured multiple hospital visits and several diagnostic and therapeutic tests. The emotional impact of these procedures should be considered, and patients offered the most agreeable environment possible. The administration of premedication prior to admission to the operating theatre must be assessed on an individual basis.
The minimum dose of whichever drug is chosen for premedication should be slowly administered, in a fractionated form if possible. This will assist with keeping the patient calm and avoid complicating airway management due to respiratory depression.
Once premedication has been administered, the patient should be monitored with a pulse oximeter and supervised.
Anaesthetic induction
When dealing with a difficult airway, the ideal scenario is to have intravenous access prior to anaesthetic induction. Usually, patients come to the operating room with venous access (some with a Port-a-Cath), but some patients may not have venous access, so the first thing to do when entering the operating room is to gain access.
Securing an intravenous line can be difficult in patients that are nervous or wary after previous difficult experiences and/or cognitively impaired.
Subject to the individual risks/benefits of each patient, premedication with nitrous oxide (when there is no pulmonary hypertension) can be administered prior to venous cannulation. Alternatively, a gradual inhalation induction with sevoflurane, using ventilatory support modes through the face mask and oxygen therapy can be used.
Airway management
As with any patient with a difficult airway, anaesthetic induction must be performed by two anaesthesiologists; in addition, there must be a main plan (both drugs and devices) and alternative plans in case of complications. These plans must be explained to all operating theatre staff and an ENT specialist must be present (to explore the neck beforehand due to the difficulty of access) in case the patient requires an emergency tracheostomy.
It should be remembered that airway involvement is progressive; thus, in younger patients, management will likely be less complicated than in older patients.
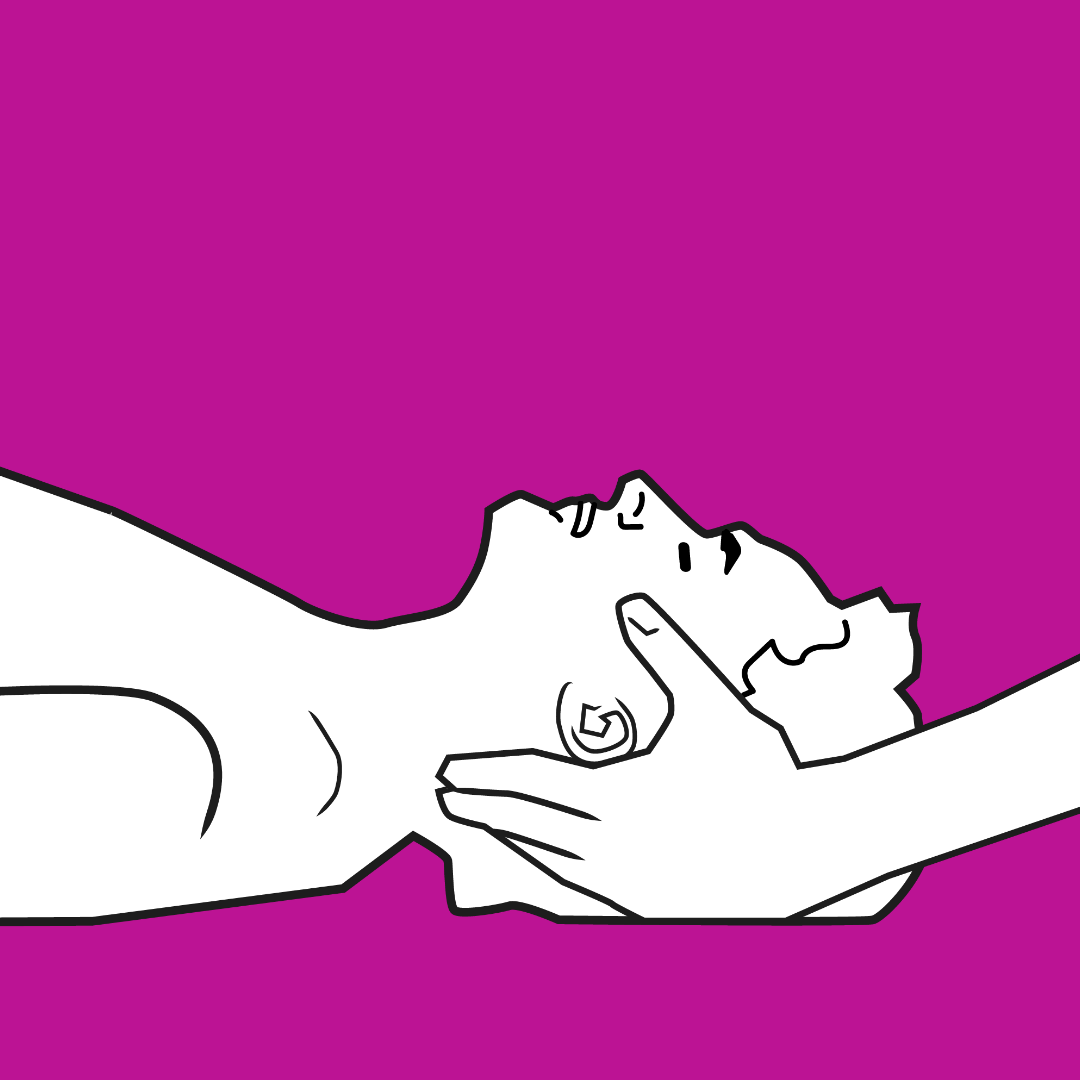
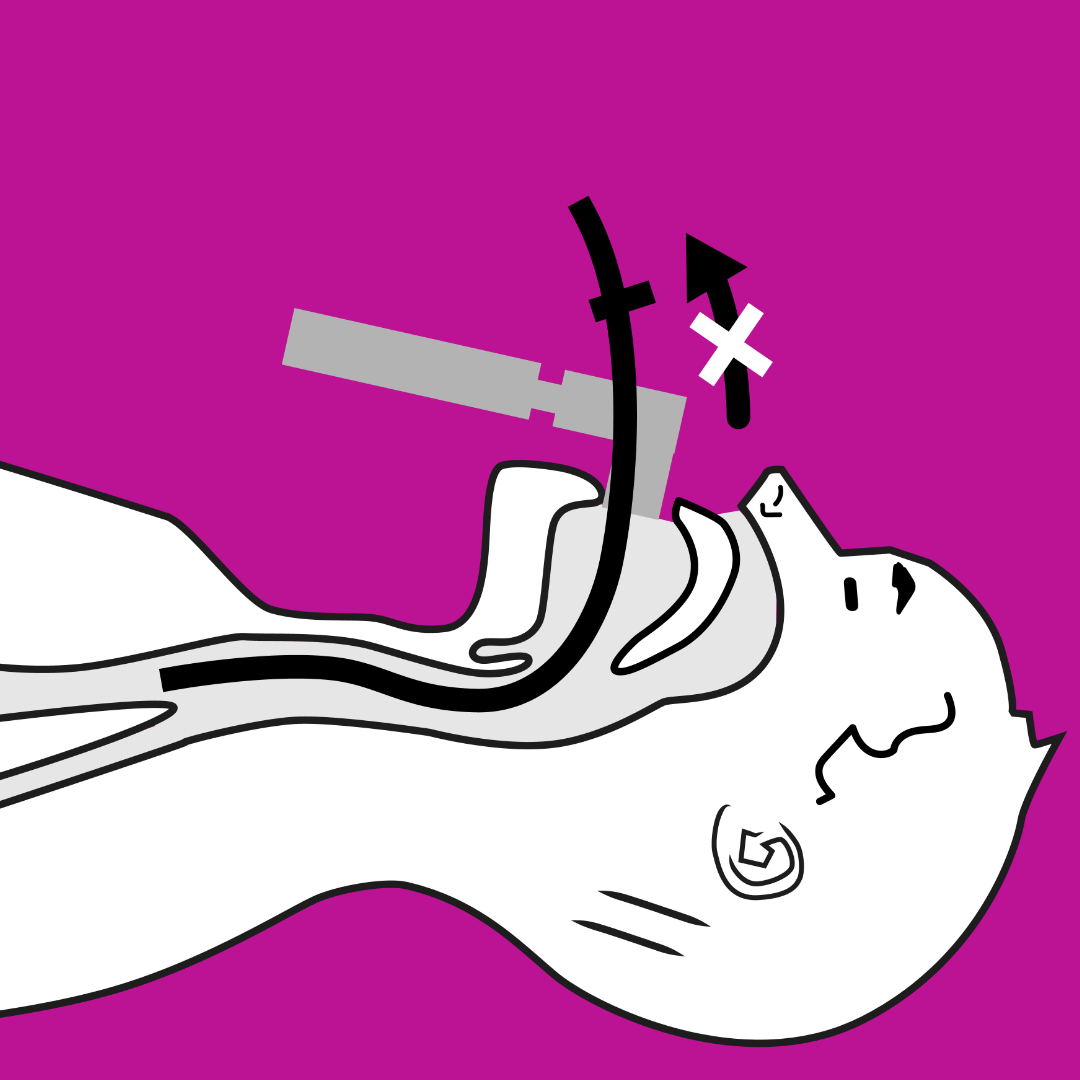
Due to the high incidence of cervical spine compression and instability (which must be well assessed in the preoperative period), manoeuvres that affect cervical mobility should be performed cautiously, avoiding hyperextension and ensuring correct protection of the cervical spine at all times.
The algorithm of action in a difficult airway must always be considered, and as these algorithms dictate, the patient’s own spontaneous ventilation must never be lost. This is vitally important in patients with MPS, as the pharynx and larynx may be so affected by GAG deposition, that the only indicator of which air passage to enter are the secretion bubbles created with each breath.
Orotracheal intubation vs. laryngeal mask
As usual, the use of devices must be individualised to the patient, the surgery and its duration.
There are several surgeries where airway management with a laryngeal mask can be performed. Even so, a bronchoscope or videolaryngoscope should be kept ready in the operating theatre in case of complications.
Once the laryngeal mask is in place, it may be advisable to pass the bronchoscope through it to check the status of the larynx in case intubation is required precipitously.
Drugs
Obtaining an adequate anaesthetic plane for the initiation of intubation manoeuvres can be achieved either intravenously or by inhalation. In both cases, the doses should be administered carefully and progressively, so that, in the event of any complication the patient can be awakened quickly and safely.
Endovenous:
There are several options depending on the experience of each anaesthesiologist and the characteristics of the patient:
Propofol
Remifentanil (as a single drug or in combination with propofol, to administer a lower dose of both drugs)
Dexmedetomidine (atrioventricular block must be ruled out beforehand)
Inhalation:
Sevoflurane: progressively until the appropriate plane is achieved
Oxygen administration must be maintained throughout the procedure, either by face mask or nasal cannulae.
A commercially available face mask with an additional valved orifice for bronchoscope access minimises air leakage, ensuring oxygen delivery throughout the intubation manoeuvre.
If such a face mask is not available, an alternative solution is to place a small endotracheal tube through a nostril into the pharynx and connect it to the ventilator. In this way, oxygen can be administered, and sevoflurane can be continued during the bronchoscopic intubation manoeuvre. Remember that with this solution, sevoflurane is not absorbed by the ventilator and will be present in the operating room environment.
Another option would be to perform bronchoscope intubation through a laryngeal mask.
In addition to the drugs used to sedate the patient, the use of topical anaesthesia on the airway, from the base of the tongue to the trachea, is essential. This prevents stimulus of the pharyngeal or laryngeal mucosa, which can provoke coughing spells, or even laryngospasm or bronchospasm.
Topical anaesthesia can be on an awake patient if minimally cooperative, or with the patient sedated.
Difficult airway devices
The main device for a difficult airway remains the bronchoscope. In recent years, the use of video laryngoscopes has become more popular due to its success in difficult airway situations, but the bronchoscope remains the device of choice.
During the intubation manoeuvre, local anaesthetic can be administered by spraying it through the working channel of the bronchoscope.
It is important to assess the situation from the beginning of the manoeuvre in the mouth. The pharyngeal and laryngeal anatomy can be very distorted, and the structures observed during the introduction of the bronchoscope can be misinterpreted.
Due to the narrowness of the patient’s airway, care should be taken to ensure that the size of the orotracheal tube is somewhat smaller than that corresponding to the patient’s age.
Ventilation
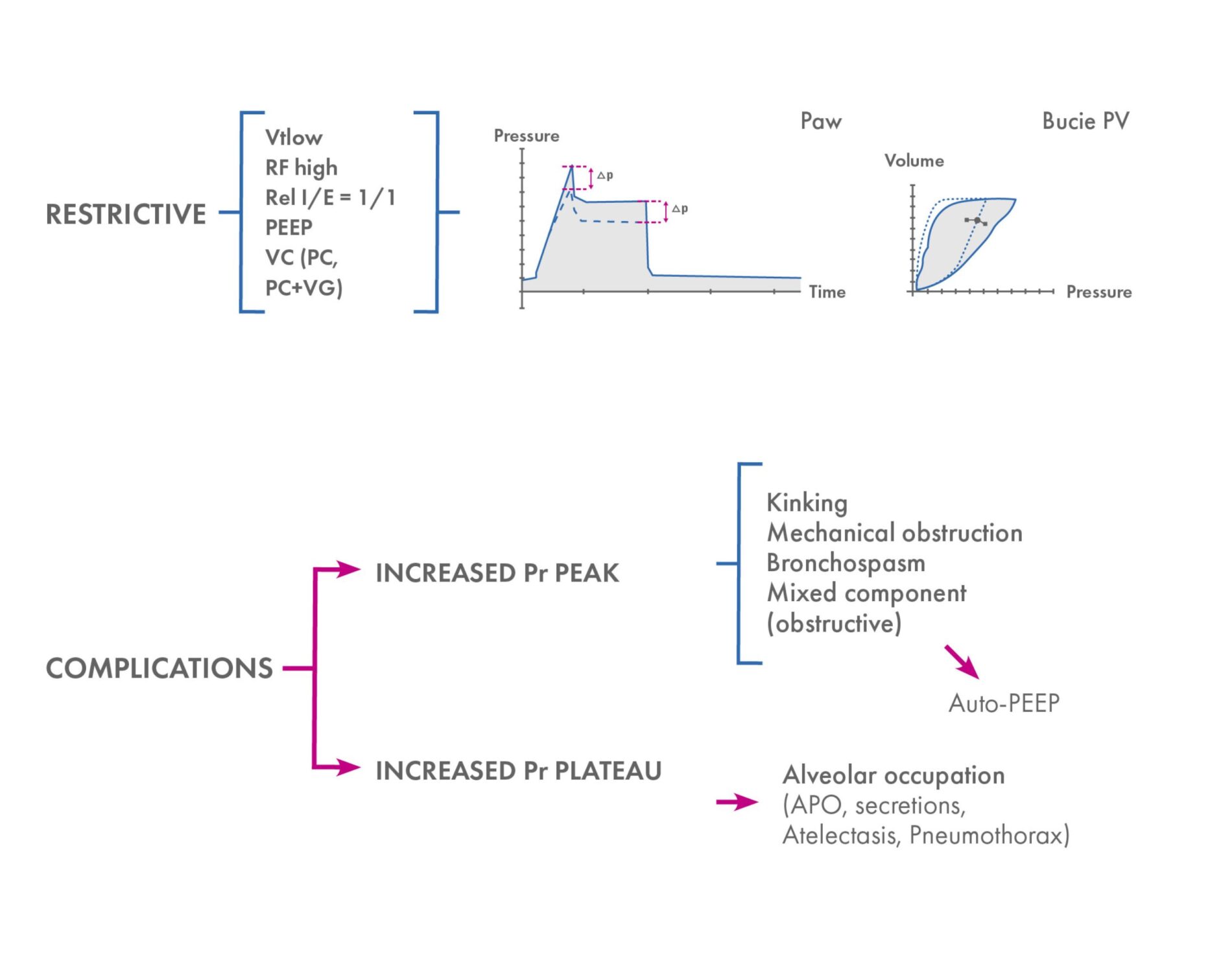
When programming the ventilator, it is important to remember that you will usually be dealing with a restrictive pulmonary pattern. An inspiration/expiration ratio closer to one should be set, analysing the possible air trapping in advanced cases, which is where it could coexist with an obstructive pattern.
MPS patients usually have elevated intrapulmonary and airway pressures, with respiratory conditions that may vary during surgery.
The usual presence of secretions in the bronchial tree must be considered, which may require aspiration through the endotracheal tube.
Ventilatory mode
Any ventilatory mode can be used depending on the patient’s condition at the time. If more information on the patient’s pulmonary status is desired, the use of volume control (VC) is useful, as this provides the plateau pressure indicating intrapulmonary pressure. If the patient requires high pressures for proper ventilation, these can be adjusted by using the pressure control (PC) or even PC + guaranteed volume (VG) mode (AutoFlow depending on the ventilator), although some of the information is lost.
Interpretation not only of pressure/time and flow/time graphs, but also of pressure/time loops is useful for early and detailed detection of possible respiratory changes or complications during surgery.
Summary of respiratory management and intraoperative respiratory complications
As these are patients with possible cardiac involvement, remember that during anaesthesia, the cardiopulmonary system functions as a unit, and changes in ventilation may affect cardiovascular function and vice versa.
For example, a decrease in exhaled CO2 can be due to respiratory or haemodynamic causes or oesophageal pressure (OP): excessive use of positive end-expiratory pressure (PEEP) can lead to alterations in cardiac function.
The possibility of cardiac involvement must always be considered even if preoperative tests do not show significant involvement.
Extubation
Extubation is a critical moment, even more so than intubation, as the airway remains equally complex. Extubating carries additional risks of oedema or bleeding caused by manipulation of the airway, surgical time and drugs administered during anaesthesia.
It is advisable to administer corticosteroids, completely reverse the effect of muscle relaxants, place a device to clear the airway and use ventilatory support after extubation.
Extubation should be performed with the patient fully awake, with active coughing and swallowing reflexes, adequate breathing and active movement and muscle strength.
Devices are available on the market to facilitate reintubation if necessary. They consist of a very thin guide that can be placed through the trachea before extubating the patient. If necessary, a tube exchanger is inserted through this guide and the patient can be intubated without all the initial manoeuvres. This fine guidewire is removed if reintubation is not required when the risk is deemed to be imperceptible. This material can be very useful in patients who are particularly difficult to intubate.
In the first minutes after extubation, ventilatory support can be administered directly from the operating room ventilator, in pressure support (PS) mode with a tightly sealed face mask. Thus, a single level of support ventilation can be prescribed (PEEP on the ventilator) or dual level (PEEP and PS), remembering to modify the trigger according to the patient’s age. In this way, non-invasive ventilation (NIV) will be performed with the operating room ventilator. The patient’s own home ventilator can also be used. Once in the post-anaesthesia recovery room, it is necessary to assess whether the patient requires NIV for some time until lung function is fully recovered.
Eye protection
It must be remembered that patients with exophthalmos should be protected with eye cream and occlusion. In surgeries requiring a prone position, special care should be taken to ensure that the eyes are free of any pressure.
Neuropathies and skeletal disorders
Due to skeletal deformities, the position during surgery may cause postoperative pain or even nerve compression. Where permitted, it is recommended that the patient (or family) explains which position he or she rests or sleeps comfortably in so they may be positioned prior to anaesthetic induction.
Skeletal deformities can complicate the performance of peripheral nerve blocks due to the modified route that the nerves may take.
Albert Sánchez Vega (2021) ‘Consideraciones anestésicas en pacientes con mucopolisacaridosis’, in Del Toro Riera, M. (ed.) Aproximación quirúrgica al paciente con mucopolisacaridosis. España: Content Ed Net, pp. 80-88.
THE CONTENT AVAILABLE THROUGH THIS PAGE IS FOR INFORMATIONAL AND EDUCATIONAL PURPOSES ONLY AND IS NOT A SUBSTITUTE LOCAL EXPERT CENTRE OR HOSPITAL MANAGEMENT PROTOCOLS. BIOMARIN DOES NOT GIVE MEDICAL ADVICE, NOR DOES IT PROVIDE MEDICAL OR DIAGNOSTIC SERVICES. YOUR RELIANCE UPON CONTENT OBTAINED BY YOU AT OR THROUGH THIS PAGE IS ON THIS UNDERSTANDING.
Register for access
Get access to the latest webinars, meetings and events about MPS; registration is quick and easy.